早稲田大学 研究
早稲田大学 研究機械学習で結晶構造の予測精度を2倍に向上
医薬品や機能性有機材料の開発を加速する新手法を開発
【注目の成果:共同研究・産学連携のためのチェックポイント】
 | 新たな有機材料の発見を加速 |
【産学連携対象 全学共通分野 Discovery Saga】
【Sagaキーワード】
ニューラルネットワーク/ワークフロー/機械学習/最適化/対称性/量子化/計算機シミュレーション/相転移/太陽/分子構造/量子化学/量子化学計算/ディスプレイ/有機エレクトロニクス/有機太陽電池/有機半導体/分子性固体/有機分子/ACT/ファンデルワールス力/電子デバイス/有機EL/有機材料/ボトルネック/太陽電池/電気伝導/電子状態/電池/有機結晶/電気伝導性/シミュレーション/ニューラルネット/機能性材料/結晶化/多孔質/多孔質材料/半導体/インフォマティクス/構造予測/機能性/結晶構造/コンフォメーション/医薬品開発
発表のポイント
機械学習を用いて、有機分子の結晶構造予測(CSP)における探索空間を絞り込む新手法「SPaDe(スペード)-CSP」を開発しました。本手法は、予測の初期段階で機械学習を用いて有望な空間群と結晶密度の候補を絞り込んだ後、結晶構造の最適化計算に高速かつ高精度なニューラルネットワークポテンシャルを用いることで、探索効率化と計算コストの大幅削減を実現しました。
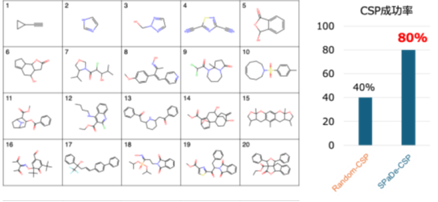
本手法を適用した実験において、有機分子の結晶構造を80%という高い成功率で予測することに成功しました。この成功率は、従来型のランダムな探索手法と比較して2倍に相当します。
医薬品開発や、有機半導体といった機能性材料の設計など、精密な結晶構造の制御が求められる分野への貢献が期待されます。
発表概要
早稲田大学データ科学センターの谷口卓也(たにぐちたくや)准教授、同大学大学院先進理工学研究科一貫制博士課程3年の深澤亮(ふかさわりょう)らの研究グループ(以下、本研究グループ)は、このたび機械学習を使って有機分子の結晶構造予測※1の成功率を向上させることに成功しました。有機分子の結晶構造を予測することは医薬品や機能性材料の開発に重要ですが、膨大な計算コストが課題でした。本研究では、機械学習を用いて有望な結晶構造の候補を効率的に絞り込み、ニューラルネットワークポテンシャル※2で高速に構造を最適化する新しい結晶構造予測ワークフロー「SPaDe-CSP」を開発しました。本手法を20種類の有機結晶に適用した結果、有機分子の結晶構造を80%という高い成功率で予測することに成功しました。この成功率は従来型のランダムな探索手法と比較して2倍に相当します。本成果は、新たな有機材料の発見を加速させるものです。
本研究成果は、英国の王立科学会が発行する「Digital Discovery」誌にて2025年10月13日(月)にオンライン公開されました。
これまでの研究で分かっていたこと
有機分子の結晶構造は、医薬品の溶解性や安定性、あるいは有機半導体の電気伝導性といった物性に直接影響を与えるため、その構造を正確に予測することは極めて重要です。しかし、有機結晶はファンデルワールス力や水素結合といった弱い分子間相互作用で形成されており、わずかなエネルギー差で多様な構造(多形)を取りうるため、最も安定な構造を予測することは非常に困難でした。従来の予測手法では、考えうる膨大な数の候補構造を生成して一つずつ評価する必要があり、その計算コストの高さが材料開発のボトルネックとなっていました。
新たに実現しようとしたこと、明らかになったこと、そのために新しく開発した手法
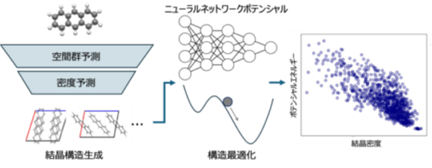
本研究では、この計算コストの問題を解決するため、機械学習とニューラルネットワークポテンシャルを組み合わせた新しい結晶構造予測※1ワークフロー「SPaDe-CSP」※3を開発しました。SPaDeとは、Space group and Packing Density predictorの略称です。分子構造を入力し、結晶の対称性を表す空間群(Space group)と、分子の詰まり具合を示す密度(Packing Density)を予測する2つの機械学習モデルを構築しました。これにより、従来の手法で多数生成されていた、密度が低く不安定な構造候補をあらかじめ排除し、有望な候補に絞って探索を行うことができます。
絞り込まれた候補構造の最適化には、量子化学計算※4に匹敵する精度をはるかに低い計算コストで実現するニューラルネットワークポテンシャル※2を用いました。このワークフローの有効性を検証するため、複雑さが異なる20種類の有機分子の結晶構造予測に挑戦したところ、80%(20種類中16種類)のケースで実験的に観測された構造を正しく予測することに成功しました(図1)。これは、機械学習による絞り込みを行わない従来型のランダム探索に比べて約2倍の高い成功率に相当し、本手法の有効性を実証するものです。一方、分子量が大きく構造が複雑な分子ほど、結晶構造予測が難しい傾向にあることが明らかになりました。